


Diamond 分子結構軟體
Diamond是傑出的分子和晶體結構顯示軟體.它結合了多種功能,可以用於含有晶體結構數據的工作,適用於教育,科學研究以及出版。Diamond像其它的軟體一樣不僅可以畫出精密的分子和晶體結構圖片,它還有一系列拓展的功能,它可以讓你很容易的從一套基本結構參數(晶胞,空間群和原子的位置)中模擬任意部分的晶體結構。
Diamond is our outstanding molecular and crystal structure visualization software. It integrates a multitude of functions, which overcome the work with crystal structure data - in research and education as well as for publications and presentations.
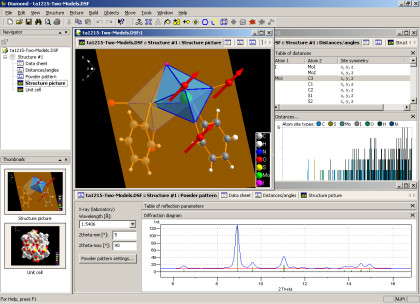
Diamond does not only draw nice pictures of molecular and crystal structures like most of its competitive programs do. It offers an extensive set of functions that let you easily model any arbitrary portion of a crystal structure from a basic set of structural parameters (cell, space group, atomic positions).
With its high data capacity, its wide range of functions beginning with the generation of molecules reaching up to the construction of rather complicated inorganic structural frameworks, Diamond is a comprehensive tool for both molecular and solid state chemists as well as for surface and material scientists.
Diamond 3 Function List
- 32 Bit MS Windows application with Multiple Document Interface (MDI), object-oriented menus, toolbars and local popup-menus. Allows 'simultaneous' handling of multiple structures.
Input and Output:
- Proprietary binary Diamond 3 Document format (extension .diamdoc) :
- Supports both crystal and molecular structures (i.e. with and without translational symmetry).
- Storage of multiple structure data sets in a document , each with:
- atomic parameters,
- cell parameters and space-group (optional),
- anisotropic displacement parameters,
- chemical and bibliographic data (author, reference, database origin, etc.).
- Supports multiple structure pictures for a structure data set. Saves your own built-up and designed frameworks of crystal structures.
- Compatible with Diamond 2 format (DSF).
- Number of atoms, bonds, polyhedra etc. limited only by RAM.
- Manual input or update of chemical, crystallographic, and bibliographic data.
- Automatic import from data formats:
- CRYSTIN download format created by ICSD or CRYSTMET
- Cambridge Structural Database FDAT format.
- Brookhaven Protein Data Bank format.
- SHELX-93 format.
- Crystallographic Information File (CIF).
- XYZ format (free format with cartesian coordinates),
- SYBYL MOL and MOL2 format,
- Cerius2 (CSSR) format,
- MDL MOL format.
- Export of structure data to:
- CIF,
- SCHAKAL,
- XYZ format.
- POV-Ray assistant to create photo-realistic scenes with shadows, reflections, textures, background graphics, and more.
- Export of structure picture's 3D world to VRML.
- Export of structure picture's 2D graphics (for post-processing e.g. in a word processor or graphics application):
- as Windows metafile (WMF, vector-oriented),
- as bitmap (BMP; width, height and resolution user-defined),
- as GIF, JPG, or PNG file, e.g. to link with an HTML document.
- Cut, Copy, or Paste of data sets between documents (together with associated structure pictures). Enables creation of small "databases".
- Search for chemical, crystallographic, or bibliographic data.
- Configurable table of data sets in a document.
- Thumbnail overview of structure pictures of a selected data set or of the whole document.
- Data sheet for comprehensive, data brief for compact and configurable textual representation of structure data.
- Printing of selected datasets, data sheet/brief, tables, or structure pictures. Textual copy of datasets via Windows clipboard for post-processing.
- Export of data sheet/brief and tables as HTML.
Construction:
- Optional assistant that helps to create a structure picture from scratch or to modify a picture.
- Optional "Auto-Builder" that creates pictures automatically or according to a user-defined strategy ("scheme"). Useful when visualizing a lot of similar structures.
- Conversion between "crystal" and "molecular" structures, i.e. adding or removal of cell and symmetry information.
- Filling of unit cell, multiple cells, any cell range, or boxes or spheres around selected central atoms.
- Filling of user-defined rectangular areas within the screen.
- Filling of slabs along a plane (hkl or least-squares) or between a plane and the walls of the coordinate system.
- Selection of atoms to construct sublattices.
- Discussion of connectivity assisted by histograms showing the distribution of distances between selected atom types, together with automatic calculation and checking of distance ranges.
- Creation of bonds automatically, basing on connecitivity, or manually by inserting bonds between two atoms each.
- Generation of atoms from parameter list serving as initial atoms for building up complex frameworks.
- Completion of coordination spheres around selected atoms.
- Automatic generation of molecules or completion of fragments which has been clipped at cell edges.
- Creation of "broken-off" bonds to signal infinitesimal chains, layers, or 3D-frameworks.
- Cut, copy and paste of structural parts between structure pictures:
- A fragment of a structure picture (or the whole picture) can be copied.
- The copied fragment can be pasted into a blank or another picture of the same data set.
- User-controlled dismantling of built-up frameworks.
- Multiple-step Undo and Redo function to enable safe experimentation with even high-complicated and unknown structural frameworks.
Visualization:
- Layout modes:
- Regular/window,
- for printout, e.g. A4 page size with white background,
- for creation of a bitmap with given x and y dimension and a resolution in dpi.
- Variable zoom factor (enhances "Page view" mode of Diamond 2).
- Models, assigned globally or individually to single or groups of atoms (allows mixing of different models in one and the same picture):
- Ball-and-stick (regular),
- ellipsoid,
- space-filling,
- sticks or wires (depending on bond radius).
- Definition of views along special axes or toward special planes.
- Central or parallel projection, depth cueing, and stereo display.
- Photorealistic rendered models with user-defined light source and material properties (OpenGL).
- Variation of colors, styles and radii of atom groups and bonds. Individual design of each single atom is possible.
- ORTEP-like atom styles (ellipses, octants) in both flat and rendering mode.
- Optionally fragmentated and two-colored bonds.
- Labelling of atoms and bonds. User-defined text, can be placed at arbitrary position of picture.
- Generation of coordination polyhedra:
- Around central atoms of selected groups or around individually selected atoms,
- built up from selected ligand atoms,
- optionally with transparent or hatched surfaces.
- Definition of (transparent) lattice planes and (best) planes or lines through selected atoms.
- Adding of vectors to atoms to indicate e.g. a magnetic moment.
- Generation of H-bonds .
- Alternative color differentation to visualize oxidation numbers, site occupation factors etc.
Animation:
- Movement of structure picture:
- Modes:
- Rotation along x-, y-, and/or z-axis,
- horizontal and/or vertical shift within drawing area,
- variation of enlargement factor (from Angstroems to centimeters),
- variation of camera distance (perspective impression).
- Controlled by:
- Mouse (the faster the mouse the faster the rotation etc.),
- keyboard (e.g. one degree rotation per keystroke),
- numerically (input through dialog).
- Optional "Spin" function, i.e. acceleration of movement.
- Continuous movement, which can be interrupted and continued.
- Modes:
- Walk-through mode, enabling the camera/viewer to navigate through the structure picture.
- Recorder that helps to create video sequences, e.g. as AVI files.
Exploration:
- Calculation of powder pattern:
- Variation of diffraction parameters:
- Radiation type: X-ray (laboratory, synchroton), neutron, electron,
- wavelength,
- LP correction,
- 2theta range,
- optional profile functions.
- Diffraction diagram (styles, colors and line weights can be configured).
- Table of reflection parameters with zoom in/zoom out and tracking through 2theta range.
- Variation of diffraction parameters:
- Calculation of distances and angles (incl. standard uncertainties):
- in a configurable table, for selected atom types and a sizeable distances range,
- around the atom(s) currently selected in structure picture.
- Graphical representation of distances as histogram with color-coded distances.
- Measuring of distances, angles, and torsion angles interactively (incl. standard uncertainties).
- Measuring of extended geometric features (incl. standard uncertainties):
- Angle between two planes (by hkl or (best) plane through 3 or more atoms),
- angle between two lines,
- angle between a normal of a plane and a line,
- distances of atoms from a plane or a line,
- centroid of a set of atoms,
- planarity or linearity of a set of atoms (distances of constituent atoms from plane/line).
- New Properties pane, displays information about:
- Contents of the structure picture (how many created atoms, bonds, polyhedra, etc.),
- the current "formula sum", that means the number of created atoms associated to atom groups,
- Info about the object that is selected in the structure picture or in the (optional) table above the properties pane, e.g. info about an atom of the parameter list,
- Table of the currently selected objects,
- Distances around the selected atom(s),
- Distances between the selected atoms,
- The center of the selected atoms (centroid),
- The planarity or linearity of the selected atoms and the deviations of the atoms from that plane or line, rsp.,
- Table of atoms assigned to the selected atom of parameter list or selected atom group,
- Table of bonds assigned to the selected bond group (i.e. atom group pair),
- Ligand, edges, and faces informations of the selected polyhedra.
System Requirements
- Personal computer with MS Windows® 98, ME, 2000, XP, or Vista (NT® 4.0 on request)
- Microsoft Internet Explorer 5.01 (or higher)
- Pentium® II compatible processor (or higher)
- 64 MByte of RAM (or more)
- Graphics resolution of 1024x768 and 16 bit color depth (or higher)
- CD-ROM drive
- Hard disk with minimum 100 MB free disk space (or more)
- Microsoft compatible mouse