


Gaussian 09 量子化學計算軟體

Gaussian 09 is up-to-date version in the electronic structural calculation program Gaussian series. It is used by the people of the new research area where the chemist, the chemical engineer, the raw chemist and there is the physical chemist and chemically an interest. Gaussian, quality of the various molecules which are induced from fundamental calculation the basic law of quantum chemistry as a foundation, and together, estimates the frequency of energy, molecular structure and molecular type. To call the short-lived intermediate field and transition state structure not only the stable molecule and to observe experimentally to difficulty or impossible chemical compound, using the reaction and the molecule under wide-ranging condition for research it is possible. Here, it introduces functional how many it is improved newly.
The reaction modeling of the enormous type which uses ONIOM
As for ONIOM, MO: It has “electronic embedding” function at the time of MM calculating. With that, it can adopt the static characteristic of MM territory when calculating the QM territory. In addition, it has the optimization algorithm which has reliability at high speed. With this algorithm, the coupling between the atoms, inside each model system it gathers at especially MM layer micro-iteration has gone between optimization steps of entire real type. With Gaussian 09, many functional strengthening below are done in regard to ONIOM:
As for ONIOM, MO: It has “electronic embedding” function at the time of MM calculating. With that, it can adopt the static characteristic of MM territory when calculating the QM territory. In addition, it has the optimization algorithm which has reliability at high speed. With this algorithm, the coupling between the atoms, inside each model system it gathers at especially MM layer micro-iteration has gone between optimization steps of entire real type. With Gaussian 09, many functional strengthening below are done in regard to ONIOM:
- Structural optimization of transition state
- From high speed IRC calculation
- The vibration calculation which includes “electronic embedding”
- Calculation in solvent
- Improvement of effective speed
- MM power place which can be customized over the whole
- It depends on accurate analytical slope and frequency, the mounting (as for parameter all customization possibilities) new AM1, PM3, PM3MM, PM6 and the PDDG semi- empirical method
Iron enzyme isopenicilin of non heme type N synthase (IPNS)
 Atom several 5,368 (as for figure hydrogen atom non indication).
Atom several 5,368 (as for figure hydrogen atom non indication).
This is typical ones of the important catalyst with basic biochemical process. It became clear the parent and the metal center of the protein how have contributed to the catalyst activity of enzyme type individually, with the modeling doing this molecule.
Reference literature below:
[M. Lundberg and T. Kawatsu and T. Vreven, M.J. Frisch and K. Morokuma and JCTC 5 (2009) 222.]
Atom several 5,368 (as for figure hydrogen atom non indication).This is typical ones of the important catalyst with basic biochemical process. It became clear the parent and the metal center of the protein how have contributed to the catalyst activity of enzyme type individually, with the modeling doing this molecule.
Reference literature below:
[M. Lundberg and T. Kawatsu and T. Vreven, M.J. Frisch and K. Morokuma and JCTC 5 (2009) 222.]
Energy plotting of the IRC calculation which utilizes ONIOM function
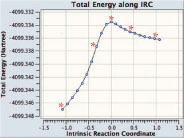 With the ONIOM function of Gaussian 09, transition state structure of the reaction which is in the midst of researching was optimized. Because completely the coupling it has done the repetition technique of macro and micro with structural optimization, the secondary coupling of QM layer of MM inside layer section is included explicitly. You verified the transition state structure which is optimized, with frequency calculation. In order and, to calculate reaction path, it made the starting structure of IRC. Plotting the result of being obtained by calculation is shown on the left.
With the ONIOM function of Gaussian 09, transition state structure of the reaction which is in the midst of researching was optimized. Because completely the coupling it has done the repetition technique of macro and micro with structural optimization, the secondary coupling of QM layer of MM inside layer section is included explicitly. You verified the transition state structure which is optimized, with frequency calculation. In order and, to calculate reaction path, it made the starting structure of IRC. Plotting the result of being obtained by calculation is shown on the left.
With the ONIOM function of Gaussian 09, transition state structure of the reaction which is in the midst of researching was optimized. Because completely the coupling it has done the repetition technique of macro and micro with structural optimization, the secondary coupling of QM layer of MM inside layer section is included explicitly. You verified the transition state structure which is optimized, with frequency calculation. In order and, to calculate reaction path, it made the starting structure of IRC. Plotting the result of being obtained by calculation is shown on the left.The structure from IRC which it calculated
The rough sketch, using GaussView 5, drew up the middle of IRC animation as the still picture. The point in regard to energy plotting which corresponds to these structures is shown, in the upper figure with the asterisk (from the left the right). Central structure is transition state. The nearby hydrogen atom of the sulphur atom which shows with yellow, it keeps spreading with reaction sutra road surface you understand.
The rough sketch, using GaussView 5, drew up the middle of IRC animation as the still picture. The point in regard to energy plotting which corresponds to these structures is shown, in the upper figure with the asterisk (from the left the right). Central structure is transition state. The nearby hydrogen atom of the sulphur atom which shows with yellow, it keeps spreading with reaction sutra road surface you understand.





Research of excitation state in vapor phase and solvent
Excitation state system and in order to research the reaction and the process it is new in Gaussian 09, many functions are included:
Excitation state system and in order to research the reaction and the process it is new in Gaussian 09, many functions are included:
- Analytical time dependence DFT (TD-DFT) slope
- EOM-CCSD method
- State specific solvation excitation and it is low transition to the level
- Franck-Condon and Herzberg-Teller analysis (FCHT)
- CIS in (equilibrium and non-equilibrium) solvent and complete correspondence of TD-DFT calculation
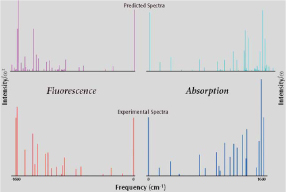
Calculated value of Qx band of porphyrin and comparison of survey spectrum.
As for these graphs, it is something which compares the high resolution semilinear absorption and emission band. Plotting strength of calculation and survey Ω (absorption) and is something which with ω3 (radiation) is divided. Ground state with DFT method excitation state structural optimization and frequency calculation were done with TD-DFT method making use of PBE1PBE function (generally known PBE0). Electron transition strength with vibrational analysis to transition state used Franck-Condon Herzberg-Teller method from ground state. These all functions are mounted on Gaussian 09. Applying 0.95, it has shortened the frequency axis of the spectrum which it calculated. Concerning more detailed calculation contents, please read the literature below:
F. Santoro and A. Lami and R. Improta and V. Barone and J. Chem. Phys. 128 (2008) 224311.
As for these graphs, it is something which compares the high resolution semilinear absorption and emission band. Plotting strength of calculation and survey Ω (absorption) and is something which with ω3 (radiation) is divided. Ground state with DFT method excitation state structural optimization and frequency calculation were done with TD-DFT method making use of PBE1PBE function (generally known PBE0). Electron transition strength with vibrational analysis to transition state used Franck-Condon Herzberg-Teller method from ground state. These all functions are mounted on Gaussian 09. Applying 0.95, it has shortened the frequency axis of the spectrum which it calculated. Concerning more detailed calculation contents, please read the literature below:
F. Santoro and A. Lami and R. Improta and V. Barone and J. Chem. Phys. 128 (2008) 224311.
Other Advanced Capability
Like below Advanced Capability is introduced into Gaussian 09, to in addition to:
Like below Advanced Capability is introduced into Gaussian 09, to in addition to:
- Solvation functional substantial strengthening: With the excitation state system which you are mentioned above new function is mounted in SCRF in addition to function. With type continual plane surface electric charge, continuity, smooth characteristic and strong characteristic of the reaction place are guaranteed. In addition, also continuity of the differential regarding the position and the external perturbation place of the atom is guaranteed. As a result, (possible at the number of steps which are not different from vapor phase) structural optimization and the accurate frequency calculation whose at high speed in the solvent reliability is higher become.
- The analytical slope which is by Brueckner Doubles (BD) method
- Additional spectrum estimate function: The analytical primary super polarizability which is by DFT numerical secondary super polarizability, analytical static and dynamic Raman strength and analytical dynamic ROA strength, improvement non harmony frequency calculation.
- Population analysis of individual track
- It was based on the fragment, first stage guess and population analysis
- Improvement of operativity: Many calculation types, reliability well recalculate possibly. Retaining and reading the layer of fixing, fragment definition and ONIOM of the atom with the fragment definition and type in the molecule and the selection and the rearranging of the standard vibration which in the residue and the frequency calculation is observed, the retention and the reading and the standard vibration of post-SCF strength are included in these.
- The Advanced Capability of many DFT: Long distance revision, empirical dispersion and double hybrid functional
- The program substantial speed improvement has done everywhere. Structural optimization of the big molecule accelerates, frequency calculation is parallel and 16 times, in the IRC calculation 3 times, improvement of 2 times has done in the angle of rotation calculation.