


Q-Chem 分子密度計算軟體

Q-Chem is the up-to-date non empirical electronic structural calculation program. First principle calculation of ground state and excitation state of the molecule is made possible, many calculation technique and the tool are offered as the ab initio software package where one is integrated.
The coverage of Q-Chem is wide, can estimate following ones.
- Molecular structure
- Chemical reaction
- Molecular vibration
- Electronic spectrum
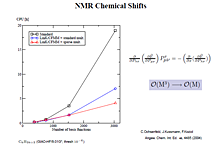
- NMR spectrum
- Solvation effect


The new function of Q-Chem 3.2:
- The DFT functional option which is added anew:
- Long distance exchange interaction revision (LRC) functional
- Baer-Neuhauser-Livshits (BNL) functional
- Variation of ωB97 functional
- Restraint conditionality DFT (CDFT)
- The empirical dispersion revision which is by Grimme
- Solvation model
- SM8 model for hydration and organic solvent (energy calculation analytical slope)
- Update of Onsager reaction place model
- Analysis of interaction between molecules:
- SCF which uses the complete localization molecular track for molecular interaction (SCF-MI)
- The Roothaan step which follows to SCF-MI (Roothaan-step) revision
- Energy bisection (EDA)
- COVP for charge-transfer (Complimentary occupied-virtual pair) analysis
- BSSE which is automated (basis-set superposition error) calculation
- Analysis of electronic movement
- Relief restraint for SCF focus (Relaxed constraint) algorithm (RCA)
- G3Large basis functional system for transition metal
- New MP2 option:
- The energy due to double basis type RIMP2 method analytical slope
- O2 energy analytical slope
- The calculation method where the wave function base in order to calculate the property of excitation state efficiently is new
- SOS-CIS for excitation state (D) energy
- SOS-CIS for excitation state (D0) energy and slope
- Connection cluster method (Coupled-cluster methods):
- IP-CISD energy and slope
- EOM-IP-CCSD energy and slope
- The parallel calculation in the connection cluster method which uses OpenMP
- QM/MM method:
- QM/MM complete Hessian appraisal
- QM/MM MBH (mobile-block) hessian appraisal
- Description of MM atom which uses the Gaussian nonlocalized electron
- The vibrational analysis which uses Partial Hessian method
- Wave function analytical tool:
- Improvement of localization tracked calculation algorithm
- Dispersion multipolar analysis
- Analytical Wigner distribution function
Q-Chem hardware requirement
- Linux 32 bit version (parallel and serial)
- Linux 64 bit version (parallel and serial)
- The Linux 64 bit version for Itanium processor (parallel and the serial) * please inquire.
- Windows (XP and Vista) 32 bit versions (parallel and the serial) * it becomes the DOS console program.
- Mac Intel OSX 64 bit version (parallel and serial)
- IBM AIX 64 bit version (parallel)